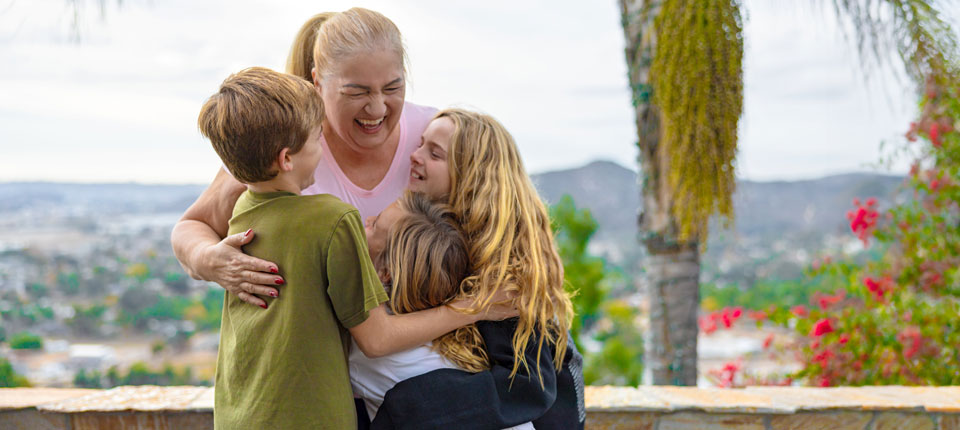Press Releases Abbott expands access to Precision Oncology portfolio through integration with Flatiron's OncoEMR® platform
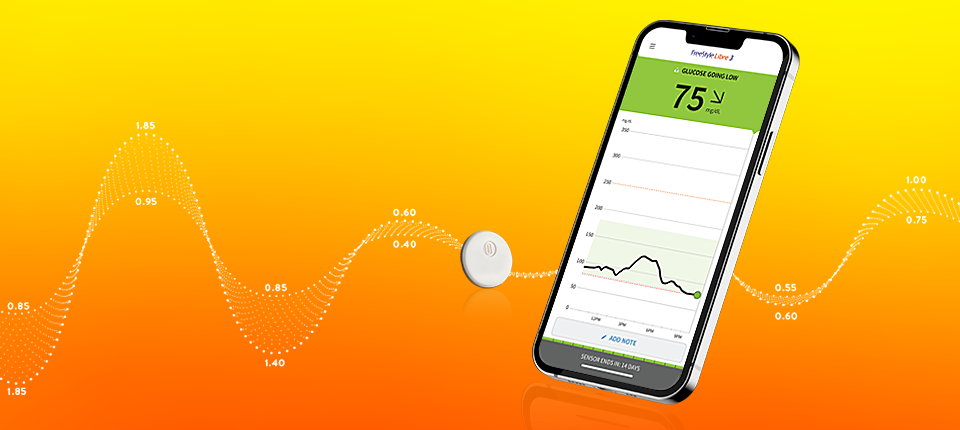
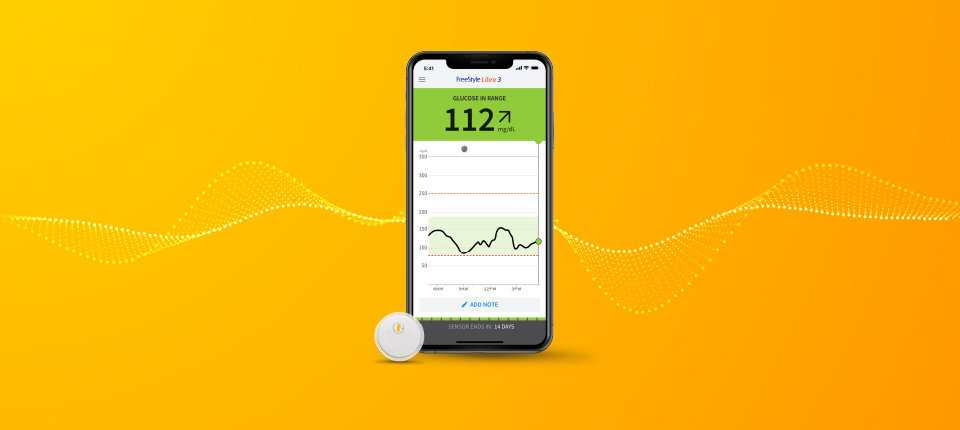
Press Releases Landmark study shows Libre technology helps people with Type 2 diabetes on basal insulin
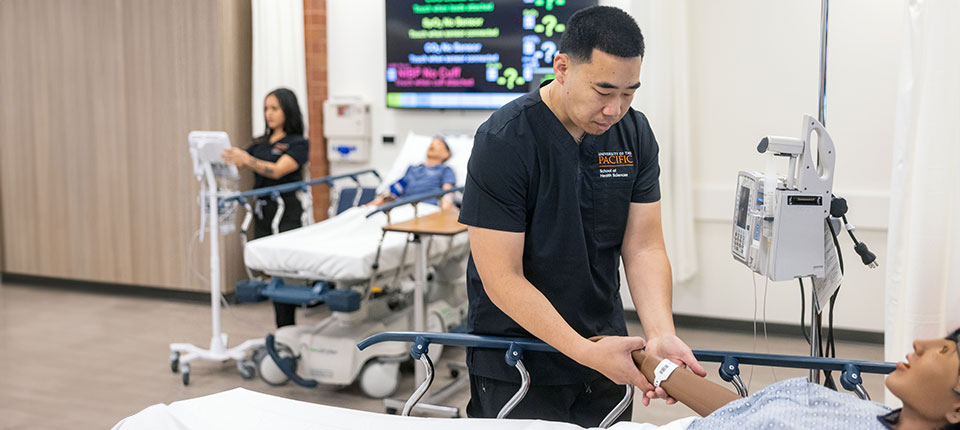
Press Releases Abbott’s next‑generation CardioMEMS™ remote heart failure monitoring reader receives FDA approval
Press Releases Late-breaking data show positive safety outcomes and closure rates for new Abbott device
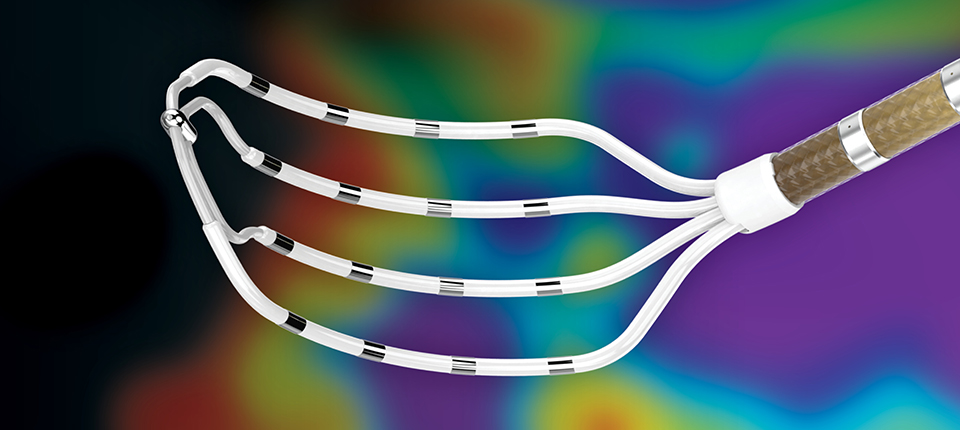
Press Releases Data highlights Abbott ablation catheters’ safety and efficacy in treating people with AFib
Press Releases Abbott Reports Fourth-Quarter and Full-Year 2025 Results; Issues 2026 Financial Outlook
Press Releases Sesame Workshop and Abbott debut new resources to help families build healthy habits early
Press Releases Abbott's Volt™ pulsed field ablation system receives FDA approval to treat patients with atrial fibrillation
Press Releases University of Wisconsin wins the ‘We Give Blood’ competition, with donations surging 319%
Press Releases Abbott initiates medical device correction for certain FreeStyle Libre® sensors in U.S.
Press Releases Abbott to acquire Exact Sciences, a leader in large and fast-growing cancer screening segment
Press Releases Abbott HeartMates community delivers holiday cheer in Hallmark Channel’s new movie ‘The More the Merrier’
Press Releases Abbott, Abbott Fund Support Northern Illinois Food Bank with $250,000 Grant to Address Childhood Hunger
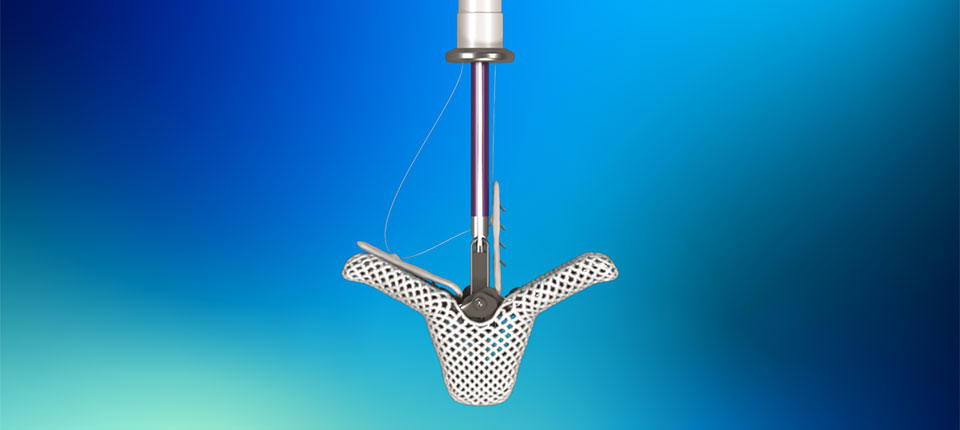
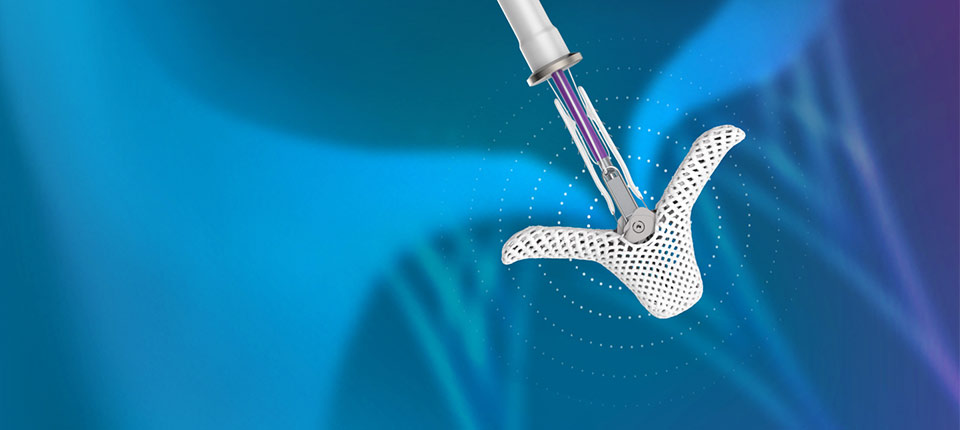
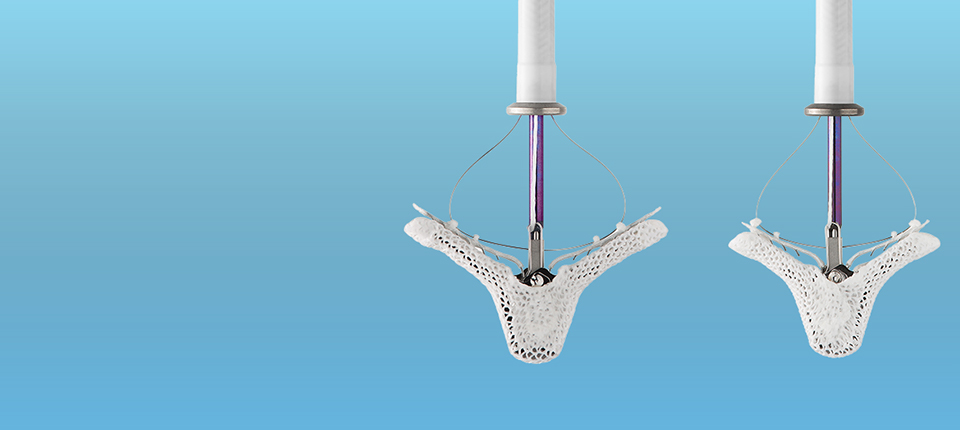
Press Releases Abbott's Navitor™ TAVI System Receives CE Mark for Expanded Indication to Treat More People With Aortic Stenosis
Press Releases Abbott's Dissolving Stent Receives CE Mark, Pioneering a New Era in Peripheral Vascular Treatment
Nutrition, Health and Wellness With infant formula, innovating means getting ever closer to breast milk
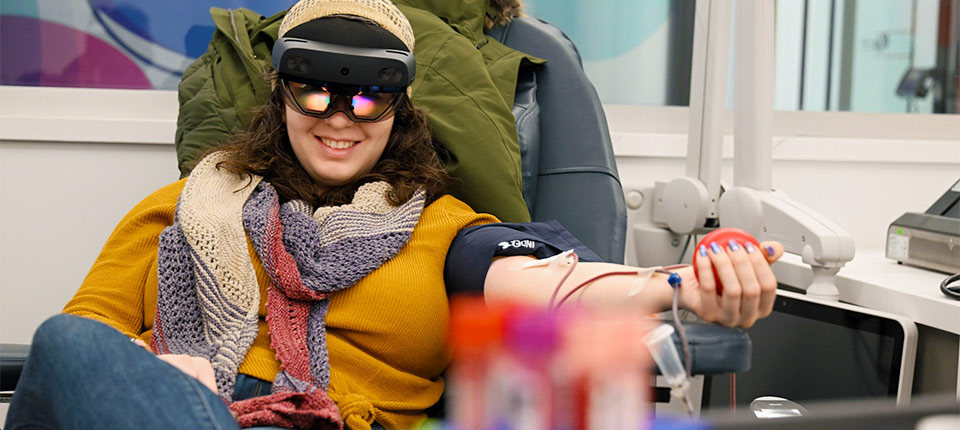
Press Releases Abbott and Big Ten Conference Rally Students, Alumni, and Fans to Save Lives and Win $1 Million for Their School by Donating Blood This College Football Season
Press Releases New Research on Abbott's Healthy Food Rx Program Shows 'Food is Medicine' Approach Helps People Living With Diabetes Eat Better and Feel Healthier
Strategy and Strength Abbott’s TriClip Offers New Treatment Option for People With Tricuspid Regurgitation
Strategy and Strength Abbott's Q3 Continues Underlying Base Business Growth Driven by Pandemic Investments
Nutrition, Health and Wellness Abbott Announces Its New Specialty and Metabolic Powder Nutritional Manufacturing Facility Will Be in Bowling Green, Ohio
Nutrition, Health and Wellness Abbott Voluntarily Recalls Certain Lots of 2 fl. oz./59 mL Bottles of Ready-to-Feed Liquid Products; Recall Is Not Expected to Impact U.S. Infant Formula Supply
Strategy and Strength Miles White to Step Down as CEO at the End of March, Remains Executive Chairman
Nutrition, Health and Wellness How Far Would You Run to Beat Cancer? Mike Sheehy Ran His Way to a World Record.
Media Contacts Find contacts at Abbott who can assist with media inquiries and press release information.
Abbott Nutrition Newsroom Our Nutrition Newsroom offers the latest nutrition-related news, research and tips.